THERAPEUTICS
BACKGROUND
The desmoplasia (fibrosis) that forms around many solid tumors is a highly complex mixture of pancreatic stellate cells, cancer associated fibroblasts, myofibroblasts, macrophages, and even endothelial cells and adipocytes. Each of these cell types secretes extracellular matrix (ECM) proteins that contribute to the complex fibrotic barrier around the tumor. Components of this fibrotic barrier include fibronectin, laminin, decorin, tenascin, periostin, multiple types of collagen and various stroma and tumor-derived enzymes.
Among the enzymes in the ECM is Quiescin Sulfhydryl Oxidase 1 (QSOX1), an over-expressed, but understudied sulfhydryl oxidase. Our group and others have reported an association of QSOX1 expression with poor outcome in several cancers including pancreatic ductal adenocarcinoma (PDAC) and luminal B breast cancer [1-7]. QSOX1 is a FAD-dependent sulfhydryl oxidase that is over-produced by many types of tumor cells and is undetectable by antibody staining in adjacent non-malignant tissues (Fig. 1)[5,8]. QSOX1 catalyzes disulfide bond formation during protein folding[9]. It is the only known enzyme that both generates and shuttles disulfides in proteins. Of note, protein disulfide isomerase (PDI) requires QSOX1 for protein folding[10] but QSOX1 does not require PDI. QSOX1 shuttles disulfides using its CxxC motifs, ultimately using FAD to transfer electrons to oxygen, producing hydrogen peroxide as a byproduct (Fig. 2). Without QSOX1 to form disulfide bonds, cysteines have free thiols which can indiscriminately cross link as they are secreted from the cell. This leads to improper disulfide cross-linking and incorrect folding of proteins, aggregate formation and a disorganized ECM.
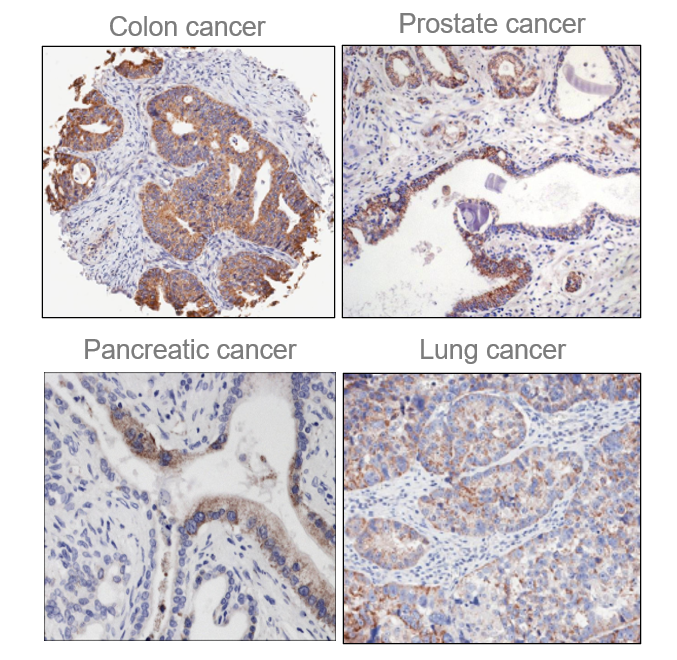
Figure 1. QSOX1 over-expression in multiple cancers. Immunohistochemistry was performed with anti-QSOX1 antibody. Brown (DBA) staining was scored 3+. Adjacent non-malignant cells and tissues do not express QSOX1.
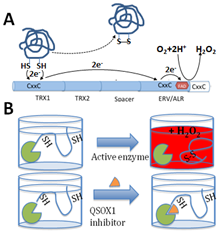
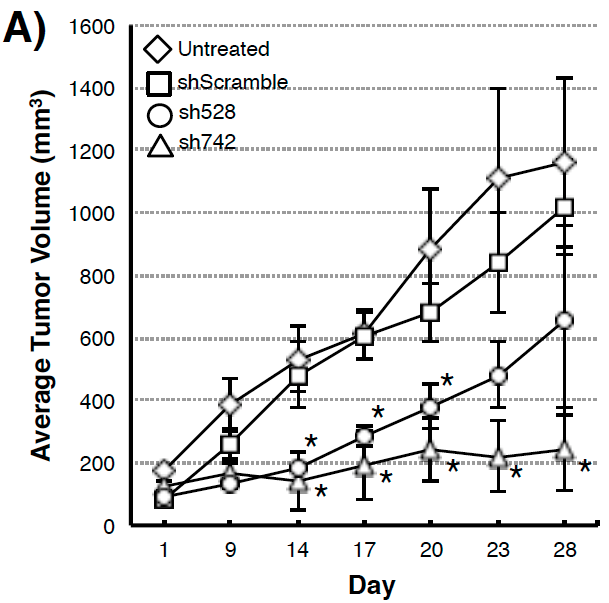
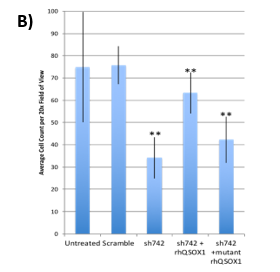
Figure 3. (A) Suppression of xenograft growth in mice of human pancreatic tumor cell line MIAPaCa2 transduced with shQSOX1 constructs sh742 and sh528. Average tumor weight is shown in the y-axis. (B) Sh742-transduced BT549 tumor cells did not invade through Matrigel-coated transwell inserts (bar 3) compared to controls (bars 1&2). Invasion was rescued by exogenous addition of functional rhQSOX1 (bar 4), but not mutated inactive rhQSOX1 (bar 5). Number of cells invading through Matrigel is shown in the y-axis.